





В данной статье приведен обзор полученных на сегодняшний день сведений, касающихся феномена эпителиально-мезенхимального перехода и его роли в развитии интерстициального фиброза почек, а также механизмов его развития и возможных точек приложения для терапевтического воздействия. Особенно интересен в этом отношении оказался трансформирующий фактор роста-β, по-видимому играющий одну из ключевых ролей в индукции эпителиально-мезенхимального перехода.
Ключевые слова: эпителиально-мезенхимальный переход, хроническая болезнь почек, фиброз почек, трансформирующий фактор роста-β.
На сегодняшний день хроническая болезнь почек (ХБП) остается важной социально-экономической проблемой, несущей за собой большие экономические потери в связи с утратой трудоспособности и инвалидизацией в молодом возрасте, значительной стоимостью лечения и реабилитации пациентов. Так, по данным регистра Российского диализного общества в 2009 году различные виды почечно-заместительной терапии получало более 24000 человек, ежегодный прирост числа этих больных в среднем составляет 10,5%.
Среди процессов, ведущих к развитию интерстициального фиброза почек и, как следствие ХБП, в настоящее время активно изучается роль эпителиально-мезенхимального перехода (ЭМП). Этот процесс возникает в результате утраты клеткой эпителиального фенотипа и приобретения ей мезенхимальных характеристик, способности активно передвигаться, изменять экспрессию адгезивных молекул и продуцировать компоненты экстрацеллюлярного матрикса (ЭЦМ).
Понятие ЭМП и его обратный процесс – мезенхимально-эпителиальный переход (МЭП) впервые были введены Элизабет Хей [11]. В настоящее время ЭМП изучается в трех аспектах (рис 1). Во-первых, установлена его роль в формировании зародышевых слоев и миграции клеток в эмбрионе позвоночных [11]. Во-вторых, ЭМП рассматривается как механизм возникновения дополнительного пула миофибробластов из эпителиальных клеток в очаге воспаления, что в патологических условиях способствует развитию фиброза органа и его недостаточности. И, в-третьих, активно обсуждается роль ЭМП в процессе метастазирования опухолей. В соответствии с этим выделяются три типа ЭМП. В данном обзоре мы рассмотрим 2 тип, принимающий участие в фиброзе почек.
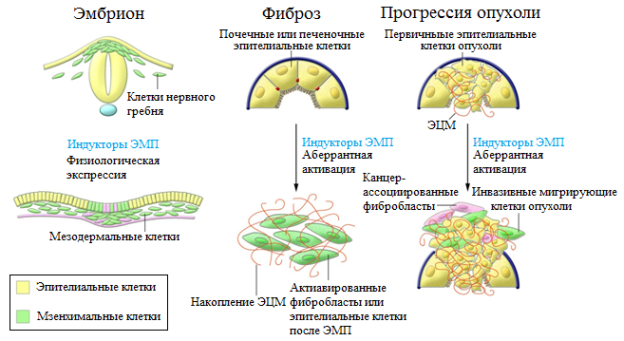 Рис. 1. ЭМП в эмбриональном развитии и при патологии (адаптировано из [2]).
Рис. 1. ЭМП в эмбриональном развитии и при патологии (адаптировано из [2]).
В норме эпителиальные клетки содержат адгезивные соединения, состоящие из Е-кадгерина вместе с катениновыми и актиновыми кольцами, плотные соединения, связанные с комплексами апикальной полярности, интегрины, взаимодействующие с компонентами базальной мембраны (рис 2а). В процессе ЭМП клетки проходят 4 условные стадии: утрачиваются свойства эпителиальной адгезии - индукторы ЭМП репрессируют транскрипцию генов, кодирующих компоненты обоих адгезивных и плотных соединений, вызывая потерю клеточной полярности /происходит деградация Е-кадгерина (рис 2б); экспрессируется De Novo α-гладкомышечный актин (α-ГМА); разрушается базальная мембрана канальцев и происходит отслаивание клеток за счет ремоделирования цитоскелета и апикального сужения (рис 2в); нарастает клеточная трансформация из эпителиального в мезенхимальный фенотип/ усиливается миграции клеток через ЭЦМ и инвазии соседних тканей за счет экспрессии рецепторов интегрина и продолжающейся активации металлопротеиназ (рис 2г) [19].Было выявлено, что контакт тубулярного эпителия с базальной мембраной стабилизирует эпителиальный фенотип, а разрыв ее основного компонента - коллагена IV типа - стимулирует ЭМП [13].
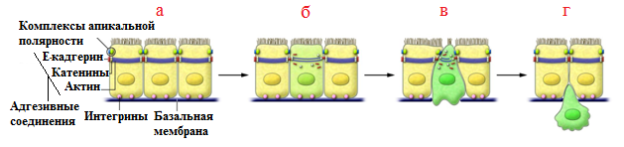 Рис. 2. Клеточные аспекты ЭМП (адаптировано из [2]).
Рис. 2. Клеточные аспекты ЭМП (адаптировано из [2]).
При ЭМП в клетке изменяется экспрессия большого числа белков, указывающих на активацию процесса – маркеры ЭМП (рис 3), и непосредственно участвующие в осуществлении программ ЭМП - его регуляторы (транскрипционные факторы, рецепторы сигнальных молекул, белки внутриклеточных сигнальных каскадов).Многие из белков, которые ранее считались лишь маркерами процесса, участвуют и в регуляции ЭМП. Так, трансфекция гена Е-кадгерина в эмбриональные фибробласты или метастатические клетки приводит к приобретению ими эпителиального фенотипа, а снижение его экспрессии может инициировать ЭМП [10].
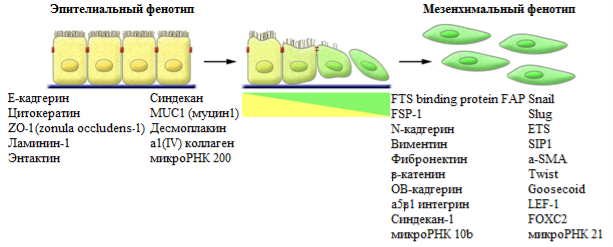
Рис. 3. Маркеры ЭМП (адаптировано из [16]).
ЭМП в патологических условиях индуцируется различными факторами микроокружения (воспалением [13] с выделением различных факторов роста, цитокинов, гипоксией, активными формами кислорода). Этот сложный многоступенчатый процесс регулируется многими системами сигнальной трансдукции с вовлечением факторов транскрипции и генными программами, регулирующими переход клеток от эпителиального фенотипа к мезенхимальному [9] (табл. 1).В отличие от 1 типа, ЭМП 2 типа может экспрессироваться в течение длительного периода времени, если воспалительный очаг не исчезает и не ослабляется [16]. Важно, что хроническое воспалительное микроокружение, общее для фиброзных и раковых клеток, выступает как решающий фактор в индукции патологического ЭМП [20].
Таблица1
ПризнакиЭМП[1].
|
Морфологические |
|
Изменение формы клеток на более вытянутую |
|
Потеря связи с соседними клетками |
|
Потеря связи с базальной мембраной и ее разрушение |
|
Молекулярные и иммуногистохимические |
|
Снижение экспрессии Е-кадгерина, ZO-1 |
|
Экспрессия виментина, N-кадгерина, фибронектина |
|
Ядерная транслокация β-катенина |
|
Нарушение межклеточных контактов |
|
Перестройка цитоскелета |
|
Изменение уровней экспрессии транскрипционных факторов, белков плотных контактов, матриксных металлопротеиназ |
|
Функциональные |
|
Способность отделяться от окружающих клеток |
|
Повышенная подвижность |
|
Повышенная инвазивность, способность разрушать базальную мембрану |
|
Устойчивость к апоптозу |
|
Устойчивость к химиотерапии |
|
Приобретение признаков раковых стволовых клеток |
|
Способность синтезировать компоненты экстрацеллюлярного матрикса |
На сегодняшний день существует несколько экспериментальных моделей фиброза почек, воспроизводимых на мышах или крысах. Модели прогрессирующего гломерулонефрита с антителами к базальной мембране капилляров клубочков [14], синдрома Альпорта [6], или спонтанного волчаночного нефрита [3], воспроизводят медленно развивающийся фиброз. Модель с односторонней обструкцией мочеточника [7], позволяет быстро добиться терминальной стадии и использовать неповрежденную почку в качестве контроля. В нашем университете на базе Центральной научно-исследовательской лаборатории успешно воспроизводилась модель односторонней обструкции почки на кроликах с 2010 по 2013 годы.
Во многих органах обычный механизм развития фиброза – увеличение количества коллаген-секретирующих миофибробластов через активацию резидентных интерстициальных фибробластов с последующим чрезмерным образованием ЭЦМ. Однако, в почках в этом процессе могут участвовать тубулярный эпителий, который претерпевает ЭMП,эндотелиальные клетки, через эндотелиально-мезенхимальный переход (ЭндМП), как показано в мышиных моделях фиброза почек [13, 29], а так же мезангиальные клетки и др. Так, около 12% фибробластов образуется из костного мозга, около 30% могут возникнуть через локальный ЭМП из эпителиальных клеток канальцев при воспалительном стрессе, и около 35% благодаря ЭндМП [15,16] (рис 4). Остальная часть возникает за счет активации фибробластов - резидентов или других мезенхимальных клеток, таких как периваскулярные гладкомышечные клетки / перициты и фиброциты [27].
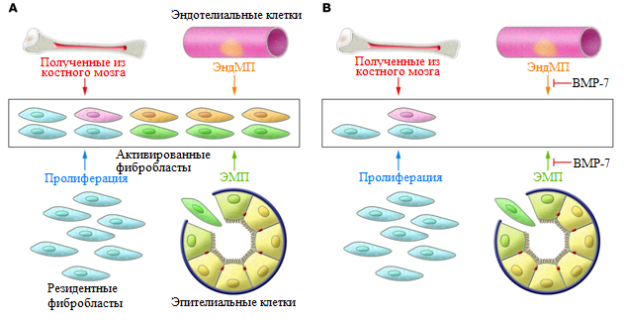
Рис. 4. А) Различные источники фибробластов при фиброзе органа. В) Системное лечение мышей с почечным фиброзом рекомбинантным человеческим ВМР-7 (адаптировано из [16]).
Прогрессирующий почечный фиброз затрагивает как клубочковый, так и тубулоинтерстициальный компоненты. Этот процесс является общим в исходе многих заболеваний, ведущих к хронической почечной недостаточности [28]. В процессе фиброза тонкая структурная организация почки утрачивается. Происходит избыточное накопление ЭЦМ. Это приводит к нарастающему снижению функции почек. Как оказалось, утрата почечной функции больше коррелирует со степенью интерстициального фиброза и атрофией канальцев, чем со степенью гломерулосклероза [17].
Повреждение почек связано со многими воспалительными клетками, которые могут стимулировать ЭMП с помощью факторов роста, таких как трансформирующий фактор роста-β (TGF-β), эпидермальный фактор роста (EGF) и фактор роста фибробластов (FGF-2) [25].
TGF-β является наиболее сильным индуктором ЭМП в культуре клеток эпителия почечных канальцев (как и в большинстве других типах клеток). Его изоформы TGF-β2 и TGF-β3 принимают участие в ЭМП во время органогенеза [5], TGF-β1 индуцирует ЭМП в различных органах (в почечных канальцах, печени, альвеолярных эпителиальных клетках и др.).В модели фиброза почек, воспроизводимой на мышах, было установлено, что TGF-β, является мощным индуктором Snail, транскрипционного фактора, который в свою очередь запускает ЭМП [24]. Этого механизма оказывается достаточно, чтобы вызвать фиброз и почечную недостаточность у взрослых трансгенных мышей. Snailтак же активируется и у пациентов с почечной фиброзом [4, 20].
В исследованиях с использованием костного морфогенетического протеина-7 (BMP-7) в качестве внутриклеточного конкурента сигналов TGF-β была показана важность последнего в индукции ЭПМ и прогрессировании фиброза почек [30]. Была установлена реверсия снижения Е-кадгерина, одного из главных эпителиальных маркеров, вызванного TGF-β [30]. В мышиной модели фиброза почек было показано обращение TGF-β-индуцированного ЭМП и восстановление поврежденных структур канальцев с появлением здоровых эпителиальных клеток при системном введении рекомбинантного BMP-7 (рис 2) [30]. В культуре клеток с Циклоспорин-А-индуцированным ЭМП, где был отмечен повышенный уровень TGF-β1, ЭМП заметно ослаблялся в присутствии анти-TGF-β1 антител. [21]
Еще один важный гуморальный фактор, потенцирующий ЭМП в клетках почечных канальцев с помощью TGF-β, ангиотензин II [26]. Высказано предположение о том, что терапевтический успех при блокаде ангиотензина II обусловлен отчасти блокадой индукции ангиотензином IITGF-β [22]. В связи с этим, блокада ренин-ангиотензин-альдостероновой системы может ослабить фиброз почек путем частичного снижения взаимодействия между ангиотензином II и TGF-β.
Несмотря на большое количество работ, выполненных по изучению ЭМП, наличие Международной Ассоциации ЭМП, есть авторы, ставящие под сомнение как существование, так и роль этого процесса в развитии фиброза при ХБП [8,12,18]. W. Kriz и соавт. отмечают, что в работе M. Iwano и соавт. [13], на которую многие ссылаются, описывая ЭМП при фиброзе, имеются неточности в проведении исследования и интерпретации его результатов, что ставит под сомнение достоверность полученных данных. Так же в ряде работ не учитывались возможные влияния экспериментальных воздействий на почечный фиброз.Rаstaldi и соавт. (2000 г.) в своей работе не получили достоверных данных, указывающих на способность клеток эпителия канальцев продуцировать коллаген [23]. В своих экспериментальных исследованиях B. D. Humphreys и соавт. полностью опровергают способность эпителиоцитов в естественных условиях трансформироваться в мезенхимальные клетки и существование ЭМП, поскольку в эксперименте при исследовании гистологического материала получили отрицательные результаты [12].
Таким образом, вопрос о роли и степени участия ЭМП в фиброзе почек и сегодня остается открытым. Несмотря на противоречивые данные, ЭМП привлекает многих исследователей, так как может пролить свет на еще одно звено в патогенезе этого инвалидизирующего процесса. Кроме того, могут быть найдены новые точки приложения для ранней диагностики, профилактики и лечения.
Литература:
1. Пучинская,М. В. Эпителиально-мезенхимальный переход в норме и патологии /М.В.Пучинская // Архив патологии. - 2015. - №1. – С. 75-83.
2. Acloque, H. Epithelial-mesenchymal transitions: the importance of changing cell state in development and disease / H. Acloque, M. S. Adams, K. Fishwick // J. Clin. Invest. - 2009. - №119 (6). –Р. 1438-1449.
3. Anders, H. Murine models of renal disease: possibilities and problems in studies using mutant mice / H. Anders, D. Schlondorff // Exp. Nephrol. - 2000. - № 8. – Р.181–193.
4. Boutet, A. Snail activation disrupts tissue homeostasis and induces fibrosis in the adult kidney / A. Boutet // EMBO J. - 2006. - №25. – Р.5603-5613.
5. Camenisch, T. D. Temporal and distinct TGFbeta ligand requirements during mouse and avian endocardial cushion morphogenesis / T. D. Camenisch, D. G. Molin, A. Person // Dev. Biol. - 2002. - № 248 (1). –Р. 170–181.
6. Cosgrove, D. Collagen COL4A3 knockout: a mouse model for autosomal Alport syndrome / D. Cosgrove // Genes Dev. - 1996. - №10. – Р. 2981–2992.
7. Diamond, J. R. Mechanisms of interstitial fibrosis in obstructive nephropathy / J. R. Diamond, S. D. Ricardo, S. Klahr // Semin. Nephrol. - 1998. - №18. – Р. 594–602.
8. Fragiadaki, M. Epithelial-mesenchymal transition in renal fibrosis - evidence for and against / M. Fragiadaki, R. M. Mason // Int. J. Exp. Pathol. - 2011. - № 92(3). –Р. 143-150.
9. Gema, M. Genetic Profiling of Epithelial Cells Expressing E-Cadherin Repressors Reveals a Distinct Role for Snail, Slug, and E47 Factors in Epithelial-Mesenchymal Transition /M. Gema, E. Cubillo, D. Sarrio // Cancer Research. - 2006. - № 66. – Р. 9543-9556.
10. Gupta, P. B. Identification of selective inhibitors of cancer stem cells byhigh-throughput screening /P. B. Gupta, T. T. Onder, G. Jiang // Cell. - 2009. - № 138(4). – Р. 645-59.
11. Hay, E. D. Organization and fine structure of epithelium and mesenchyme in the developing chick embryo. In Epithelial-mesenchymal interactions. R. Fleischmajer and R.E. Billingham, editors. Williams & Wilkins. Baltimore, Maryland, USA, 1968. – Р. 31–55.
12. Humphreys, B. D. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis / B. D. Humphreys, S. L. Lin, A. Kobayashi // Am. J. Pathol. - 2010. - № 176. - Р. 85‐97.
13. Iwano, M. Evidence that fibroblasts derive from epithelium during tissue fibrosis /M. Iwano, D. Plieth, T.M. Danoff //J. Clin. Invest. - 2002. - № 110. - Р. 341-350.
14. Kalluri, R. Susceptibility to anti-glomerular basement membrane disease and Goodpasture syndrome is linked to MHC class II genes and the emergence of T cell-mediated immunity in mice / R. Kalluri, T. M. Danoff, H. Okada // J. Clin. Invest. - 1997. - № 100. – Р. 2263-2275.
15. Kalluri, R. Epithelial mesenchymal transition and its implications for fibrosis /R. Kalluri, E. G. Neilson // J. Clin. Invest. - 2003. - № 112. – Р. 1776-1784.
16. Kalluri, R. The basics of epithelial-mesenchymal transition / R. Kalluri, R. A. Weinberg // J. Clin. Invest. -2009. - № 119 (6). – Р.1420-1428.
17. Klahr, S. Progression of chronic renal disease / S. Klahr, J. Morrissey // Am. J. Kidney Dis. - 2003. - № 41 (3). – Р. 3-7.
18. Kriz, W. Epithelial-mesenchymal transition (EMT) in kidney fibrosis: fact or fantasy? / W. Kriz, B. Kaissling, M. Le Hir // J. Clin. Invest. - 2011. - 121 (2). – Р.468-474.
19. Liu, Y. Epithelial to mesenchymal transition in renal fibrogenesis: Pathologic significance, molecular mechanisms and therapeutic intervention / Y. Liu // J. Am. Soc. Nephrol. - 2004. - № 25. – Р.1‐9.
20. Lopez-Novoa, J. M. Inflammation and EMT: an alliance towards organ fibrosis and cancer progression / J. M. Lopez-Novoa, M. A. Nieto // EMBO Mol. Med. - 2009. - № 1. (6). – Р. 303-314.
21. McMorrow, T. Cyclosporine A induced epithelial–mesenchymal transition in human renal proximal tubular epithelial cells /T. McMorrow, M. M. Gaffney, C. Slattery // Nephrol. Dial. Transplant. - 2005. - № 20. – Р. 2215–2225.
22. Peters, H. Targeting TGF-beta overexpression in renal disease: maximizing thе antifibrotic action of angiotensin II blockade / H. Peters, W. A. Border, N. A. Noble // Kidney Int. - 1998. - № 54 (5). –Р. 1570-1580.
23. Rastaldi, M. P. Epithelial‐mesenchymal transition of tubular epithelial cells in human renal biopsies / M. P. Rastaldi, F. Ferrario, L. Giardino // Kidney Int. - 2002. - № 62. – Р. 137‐146.
24. Sato, M. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction / M. Sato, Y. Muragaki, S. Saika // J. Clin. Invest. - 2003. - № 112. – Р.1486-1494.
25. Strutz, F. Role of basic fibroblast growth factor-2 in epithelial-mesenchymal transformation / F. Strutz // Kidney Int. - 2002. - № 61. – Р.1714-1728.
26. Yang, J. Hepatocyte growth factor gene therapy and angiotensin II blockade synergistically attenuate renal interstitial fibrosis in mice / J. Yang, C. Dai, Y. Liu // J. Am. Soc. Nephrol. - 2002. - № 13(10). – Р. 2464-2477.
27. Zavadil, J. TGF-beta and epithelial-to-mesenchymal transitions / J. Zavadil, E.P. Bottinger // Oncogene. - 2005. - № 24. – Р. 5764-5774.
28. Zeisberg, M. The role of epithelial-to-mesenchymal transition in renal fibrosis /M. Zeisberg, R. Kalluri // J. Mol. Med. - 2004. - № 82(3). – Р.175-181.
29. Zeisberg, E.M. Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition / E. M. Zeisberg, S. E. Potenta, H. Sugimoto // J. Am Soc. Nephrol. - 2008. - №19. – Р. 2282-2287.
30. Zeisberg, E.M. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury / E. M. Zeisberg // Nat. Med. - 2003. - № 9.- Р. 964-968.
