





Филадельфийская хромосома (Ph-chromosome) является наиболее частой цитогенетической аномалией, обнаруживающейся в зрелых В-клетках пациентов с хроническим миелоидным лейкозом (ХМЛ), причем встречаемость этой мутации растет с возрастом, у детей выявляясь в 5 %, а у взрослых — в 50–90 %, достигая максимума у больных в возрасте 35–50 лет [1].
В данном обзоре обобщены результаты работ, опубликованных в системе PubMed и посвященных механизму формирования данной генетической аномалии, а также кратко приведены последствия ее возникновения.
Ключевые слова: филадельфийская хромосома, транслокация, хронический миелолейкоз
Филадельфийская хромосома была открыта в 1960 г учеными из Университета Пенсильвании в Филадельфии (за что и была так названа), в 1973 г в работе Rowley [цит. по 2] было доказано, что она образуется именно путем транслокации. Так, у всех пациентов с ХМЛ в исследовании была обнаружена дополнительная последовательность на длинном плече 9 хромосомы. Это позволило предположить, что Ph-хромосома получается путем переноса части 22ой хромосомы на 9ую (а не простой делеции 22ой хромосомы, как думали изначально) [2].
Транслокация приводит к тому, что участок с онкогеном c-abl, кодирующим тирозинкиназу, переносится к специфическому участку разрыва на 22 хромосоме (breakpoint cluster region — bcr), вместе с тем индуцируя перенос 3'-части гена Абельсона (Abl) с 9ой хромосомы на 22ую вблизи 5'-конца такого же участка bcr [3]. Наиболее важная часть всего процесса — перестановка гена Abl на 22 хромосому рядом с геном bcr и образование химерного гена BCR-ABL [4].
Доказательство и основание переноса гена abl на 22ую хромосому приводятся ниже по работе [5]. Последовательность человеческого гена c-abl имеет гомологию с трансформирующим компонентом вируса мышиного лейкоза Абельсона (A-MuLV). Это ретровирус, имеющий геном в виде одноцепочечной РНК и способный встраиваться в геном клетки-хозяина путем образования ДНК с помощью обратной транскриптазы. Основной продукт A-MuLV –гена — полипротеин р120gag-abl, обладающий тирозинкиназной активностью (как и продукт химерного человеческого гена). Таким образом, можно говорить о трансформирующей и «транслоцирующей» способности самого гена, возможно, объясняющихся внедрением части вирусного генома в человеческий.
Что касается переноса именно между этими двумя хромосомами, здесь все не так однозначно. Одним из методов диагностики ХМЛ является автоматизированное кариотипирование клеток в стадии метафазы [6], на которой и выявляются атипичные хромосомы (см. Рис.1). Это дает основание полагать, что именно на стадии метафазы, когда хромосомы выстраиваются в экваториальной плоскости клетки, обеспечивается такое взаимное расположение 9ой и 22ой хромосомы, что возникает предпосылка к обмену их участками.
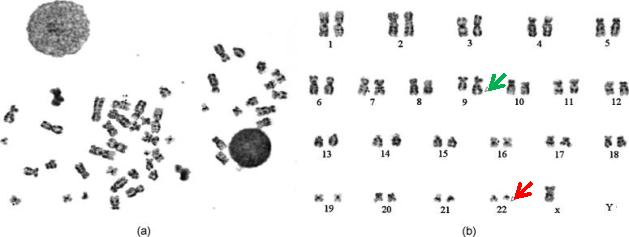
Рис. 1. а) Анализируемая клетка в метафазу и b) ее кариотип. Зеленая стрелка — удлиненная 9ая хромосома, красная стрелка — укороченная 22ая хромосома (Филадельфийская). Иллюстрация из [6] с изменениями.
Нормальный продукт гена BCR — цитозольный фосфопротеин массой 160кДа (р160BCR), функция которого не до конца определена. Известно, что первый экзон гена BCR кодирует серин/треонин-киназную активность этого белка, N-концевой домен которого отвечает за формирование димерной структуры, а С-конец обладает ГТФ-азной активностью. Также р160BCR может быть вовлечен в каскад Ras путем фосфорилирования по тирозину в 177 положении, который таким образом становится способным связывать один из адаптерных белков этого пути [4].
Нормальный продукт гена ABL — тирозинкиназа массой 145 кДа (р145ABL), участвующая в процессах клеточного деления, дифференцировки, адгезии и ответа на стрессовые воздействия. С-конец отвечает за связывание с ДНК, а также имеет сайт связывания с актином. На N-конце белка находится три Src-домена: SH3, SH2, SH1. SH3- и SH2-домены регулирующие, а SH1-домен отвечает за наличие функции тирозинкиназы. Мутации в SH2-домене ослабляют связывание фосфотирозина, что приводит к снижению функции белка. SH3-домен имеет депрессорное значение, поэтому именно его удаление превращает белок в онкопротеин, т к его тирозинкиназная активность сильно возрастает [4].
Филадельфийская транслокация дает 3 новых продукта: p190BCR-ABL, p210BCR-ABL, p230BCR-ABL, в каждом из которых нормальные последовательности р145ABL и р160BCRсоединены голова-к-хвосту [4]. Именно изоформы p210BCR-ABL обладают повышенной тирозинкиназной активностью и играют огромную роль в развитии хронического миелолейкоза.
Существует 2 изоформы p210BCR-ABL: b2a2 и b3a2, различающиеся между собой на 25 аминокислот, кодируемых экзоном b3. На настоящий момент нет данных о том, как изоформенный состав связан с различными проявлениями ХМЛ [7].
Конечный продукт химерного гена представляет собой тетрамер, олигомеризация также стимулирует тирозинкиназную активность химерного белка [4].
Помимо этого классического варианта с транслокацией t(9;22), зарегистрированы больные ХМЛ с другим кариотипом. Так, в 1970 г коллективом авторов [цит. по 2] был описан случай 45-летней женщины с типичным ХМЛ. Ей было проведено генетическое исследование, в результате которого было установлено: во всех клетках костного мозга, взятых на исследование, обнаружены филадельфийские 22ые хромосомы, однако 9ые хромосомы во всех образцах интактны (см. Рис.). Это позволило предположить, что не всегда Ph-хромосома образуется одинаково: возможны как варианты с транслокацией t(9;22), так и транслокации между другими хромосомами, а также и простое выпадение фрагментов 22ой хромосомы. Вероятно, таким разнообразием цитогенетических механизмов объясняется гетерогенность течения ХМЛ [2].
Как уже было сказано, главный момент всех перестановок — сближение фрагментов ABL и BCR, поэтому в итоге не очень важно, как именно это сближение было достигнуто. Все вышеперечисленные варианты ведут к неконтролируемому функционированию мутантного продукта, что приводит к стимуляции многих путей клеточной прогрессии, но в особенности — гемопоэтического — поражаются стволовые клетки костного мозга. В результате наблюдается избыточная продукция незрелых клеток миелоидного ряда с неконденсированным хроматином — промиелоцитов, которые «давят» все остальные ростки в костном мозге, вытесняют жировую ткань, эритроциты, тромбоциты и т. д. (доля миелоидной ткани в межбалочных пространствах костного мозга может достигать 95–100 %).
Интересно, что несмотря на то, что у пациентов с ХМЛ также может наблюдаться миелофиброз (замещение гемопоэтической ткани костного мозга фиброзной), химерный ген обнаруживается только в клетках гемопоэтической ткани красного костного мозга, и не обнаруживается в фибробластах, что говорит о том, что фиброз является вторичным процессом, не связанным с мутацией напрямую [8].
В результате действия химерного белка нарушаются пути регуляции клеточного цикла, увеличивается пролиферация клеток, их чувствительность к факторам роста, а к проапоптотическим сигналам снижается, так же, как и снижается адгезивная способность к компонентам стромы костного мозга. Все это приводит к тому, что мутантные клетки имеют преимущество в делении (т. к. не чувствительны к апоптозу и быстрее делятся) и вытесняют здоровые клоны, а также выходят в кровь (из-за потери адгезивности) [9]
Почему же эти мутации затрагивают именно гемопоэтические клетки? Возможно, дело в том, что гемопоэтические и стромальные клетки происходят от разных предшественников. В эксперименте [10] было доказано, что стромальные клетки (адипоциты, фибробласты, эндотелиальные клетки и т. п.) и гемопоэтические — это две разные линии. Соответственно, и потенциал генетических изменений у них разный. И, конечно, имеет значение, что данные клетки относятся к быстро делящимся (все ростки включают регулярно обновляемые клетки). Чем быстрее клетки должны обновляться, тем раньше станут заметны их нарушения.
Исходя из этих данных, можно объяснить симптомы ХМЛ. Усталость, истощение, лихорадка являются прямым следствием воспалительных процессов, которые сопровождают любую опухоль; геморрагии отражают тромбоцитопению; боль в костях является следствием неконтролируемой пролиферации клеток в костном мозге; гепато- и спленомегалия, а также лимфаденопатия объясняются заселением этих органов бластными формами; неврологические симптомы возникают вследствие метастазов в головной и спинной мозг. Смерть больных наступает от инфекционных процессов или геморрагического синдрома [11].
Литература:
- Forghieri F., Luppi M., Potenza L. Philadelphia chromosome-positive Acute Lymphoblastic Leukemia. Hematology, 2015; 10(20): 618–619. URL: http://dx.doi.org/10.1179/1024533215Z.000000000402
- Mitelman F.Heterogeneity of Ph1 in chronic myeloid leukaemia. Hereditas, 1974; 76(2):315–6.
- Wang Z., Ze W., Meng F., Xin X. Chronic myeloid leukemia
- with variation of translocation at (Ph) [ins (22;9) (q11;q21q34)]: a case report, Int J Clin Exp Pathol, 2015;8(10):13707–13710
- Hai A., Kizilbash N. A., Zaidi S. H. H.. Differences in structural elements of Bcr-Abl oncoprotein isoforms in Chronic Myelogenous Leukemia. Bioinformation, 2014; 10(3): 108–114. doi: 10.6026/97320630010108
- Klein A., Kessel A. G., Grosveld G., Bartram C. R., Hageimeijer A., Bootsma D., Spurr N. K., Heisterkamp N., Groffen J., Stephenson J. R. A cellular oncogene is translocated to the Philadelphia chromosome in chronic myelocytic leukaemia. Nature, 1982; 300: 765–767. [PMID: 6960256 DOI: 10.1038/300765a0]
7. Wang X., Zheng B., Li S., Mulvihill J. J., Chen X., Liu H. Automated identification of abnormal metaphase chromosome cells for the detection of chronic myeloid leukemia using microscopic images. J Biomed Opt., 2010; 15(4)046026, doi: 10.1117/1.3476336
8. Melo JV, The molecular biology of chronic myeloid leukaemia.Leukemia, 1996; 10(5): 751. PMID: 8656667
- Michiels J. J., Valster F., Wielenga J., Schelfout K., Raeve H.European vs 2015-World Health Organization clinical molecular and pathological classification of myeloproliferative neoplasms, World J Hematol, 2015; 4(3): 16–53. DOI: 10.5315/wjh.v4.i3.16, URL: https://www.researchgate.net/publication/317070261_2016_WHO_Clinical_Molecular_and_Pathological_Criteria_for_Classification_and_Staging_of_Myeloproliferative_Neoplasms_MPN_Caused_by_MPN_Driver_Mutations_in_the_JAK2_MPL_and_CALR_Genes_in_the_Context_of
- Хронические миелопролиферативные заболевания. Классификация, диагностика и лечение. Пособие для студентов IV,V,VI курсов, интернов, клинических ординаторов и врачей. СПб — Санкт-Петербургский государственный медицинский университет имени академика И. П. Павлова, 2005. -39с. URL: http://open.ifmo.ru/images/7/70/94621_metod_mielo.pdf
- — Athanasou N. A., Quinn J., Brenner M. K., Prentice H. G., Graham A., Taylor S.,Origin of marrow stromal cells and haemopoietic chimaerism following bone marrow transplantation determined by in situ hybridization, Br. J. Cancer,1990;61, 385–389
- Шиффман Ф.Дж. Патофизиология крови. Пер. с англ. — М. — СПб.: «Издательство БИНОМ» — «Невский Диалект», 2000. — 448 с., ил.
