





Ключевые слова: рентгеноспектральный флуоресцентный анализ, вязкость, проточно-пропорционального детектора, спектрометр VRA-90, внутренний стандарт, экспрессность, схеме Соллера, интенсивность фона, смесь нафтената свинца.
Введение. Среди гетеро- и микроэлементов, обнаруженных в нефти, сера играет особенно важную роль. Сернистые соединения нефти отрицательно влияют на показатели процессов нефтепереработки и нефтехимии, ухудшают качество товарных нефтепродуктов и в то же время являются потенциальным источником сырья для химической промышленности. Обязательной частью контроля качества нефти и нефтепродуктов является определение содержания серы.
Обзор зарубежных литературных источников показывает, что рентгеноспектральный флуоресцентный анализ (РСФА) эффективно используется при определении отдельных элементов в органических материалах: P,Ca, Zn, Ba в маслах [1], Pb, в бензине [2], серы и других примесей в нефти и продуктах её переработки.
Большинство нефтепродуктов представляют собой жидкости различной вязкости (нефти, нефтяные фракции, масла). Однако непосредственное применение жидких излучателей при рентгенофлуоресцентном определении легких элементов (Z<20) сдерживается необходимостью использования кювет с тонкими окнами и неудовлетворительным пределом обнаружения элементов. При работе с вакуумом такие окна кювет легко деформируются и разрываются, что может привести к выбросу жидкости в пространство спектрометрической камеры. Применение гелиевой атмосферы также является проблематичным, так как его атомы диффундируют через окно проточно-пропорционального детектора и могут вызывать изменение амплитуды зарегистрированных сигналов. Кроме этого, не все приборы имеют приспособления, необходимые для работы с гелием. Указанное выше вынуждает при подготовке жидких проб к РСФА переводить их в «сухое» состояние.
Существующие способы подготовки жидких проб весьма разнообразны.
В работе [3] для определения серы в маслах пробу кристаллизуют в парафине. Широкое распространение получил способ нанесения жидкости на бумажные фильтры. Последний способ отличается простотой и экспрессностью, но неравномерное распределение пробы по фильтру снижает точность результатов анализа, а использование небольшой аликвотной доли и разбавление целлюлозой ухудшают предел обнаружения элементов. В работе [4] для определения серы в жидком топливе пробу замораживали, а в [5] предложили переводить водные растворы в твёрдую форму, используя в качестве отвердителя желатин.
Настоящая работа посвящена разработке методики определения серы в нефтяных фракциях, используя на подготовительном этапе переведение пробы в загущенное состояние с помощью полимера.
Методика эксперимента. Как отмечалось выше, проведённый эксперимент относится к рентгенофлуоресцентным анализам, показанный в литературе [6]. Для этого были использованы фракции ДЖАРКУРГАНСКИХ нефтей, отобранные в интервале 250–500оС. Исследуемые нефти относятся к сернистым нефтям (содержание серы в сырых нефтях составляет 1,6–1,85 мас. %). Содержание серы в анализируемых материалах изменяется в диапазоне 0,3–3,5мас. %.
Предварительные исследования показали, что наилучшей совместимостью с нефтями и нефтепродуктами обладает атактический полипропилен (эфирная, гептановая, гексановая фракции), что обусловило выбор этого полимера в качестве загустителя.
Для загущения жидкую пробу смешивали в соотношении 1:4 с 30 %-ным раствором полипропилена в органическом растворителе. В зависимости от содержания серы можно брать и большее количество раствора полимера. Загущение происходит по мере испарения растворителя в сушильном шкафу при температуре 60–70оС в течение 3–5 ч. в зависимости от растворителя (гексан, гептан, толуол). Готовый излучатель представляет собой полимерную матрицу, в которой равномерно распределена анализируемая проба.
В основу методики положен способ внутреннего стандарта, позволяющий учесть влияние химического состава и качества поверхности излучателя на интенсивность Кα- линии серы (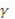 =0,537 нм.). Использование этого способа несущественно снижает экспрессность методики, так как элемент сравнения вводится в жидкую пробу. Элементом сравнения служил свинец, Мα -линия (=0,529 нм.).
=0,537 нм.). Использование этого способа несущественно снижает экспрессность методики, так как элемент сравнения вводится в жидкую пробу. Элементом сравнения служил свинец, Мα -линия (=0,529 нм.).
Интенсивность аналитических линий измеряли на рентгеновском спектрометре VRA-90 (ГДР); рентгеновская трубка с хромовым анодом; режим работы трубки-50 кВ, 16 мА: аналитические линии выделяли по схеме Соллера, коллиматор 0,40,кристалл-анализатор РЕ; детектор — проточно-пропорциональный счетчик (газовая смесь-90 % аргона, 10 % метана). Время набора импульсов 60с. Интенсивность фона измеряли рядом с линией (20=77,850). В комплект спектрометра входит компьютер.
График, построенный в координатах
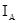 /
/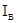 =
=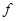 (
(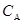 ),
),
имеет вид прямой, описываемый линейным уравнением
=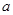 +
+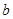 /,(1)
/,(1)
где - концентрация определяемого элемента; и– коэффициенты регрессии; /– измеренные интенсивности линий определяемого сравниваемого элементов. Коэффициенты и находили после измерения серии образцов сравнения с помощью компьютера; тогда уравнение (1) принимает вид
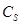 = -0,063 + 0,158∙
= -0,063 + 0,158∙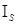 /
/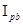 ,
,
Полученные коэффициенты остаются в памяти компьютера, что позволяет сразу после измерения интенсивности аналитических линий в образцах проб получать содержание серы.
Приготовление излучателей. В навеске пробы 1г добавляли 6 мл раствора внутреннего стандарта, представляющего собой смесь нафтената свинца и 30 %-ного раствора полипропилена в гексане; концентрация элемента сравнения в растворе пробы составит тогда 0,4 мас. %. Нефтяные фракции, выкипающие в интервале температур 380–5000С, перед отбором пробы нагревали до 600С для достижения гомогенности. После тщательного перемешивания отбирали аликвоту 2,5 мл, заливали в металлические кюветы и помещали на 3 ч в сушильный шкаф, нагретый до 600С для испарения гексана, в результате чего формировался загущенный образец-излучатель.
Образцы сравнения готовили, разбавляя различные навески дважды перекристаллизованного дибензилдисульфида (СS=26,02 %) гексадеканом (до 1г), с содержанием серы в интервале 0,3–3 мас. % и загущая так же, как указано выше.
Разработанной методикой проанализирована большая группа проб нефтяных фракций, проведены метрологические исследования методики (табл. 1).
Таблица 1
Проверка правильности определения серы в нефтяных фракциях способом добавок. n=3, доверительная вероятность Р=0,95
|
Пределы отбора фракций, ºС |
Введено, % |
|
|
|
|
|
260–270 270–280 280–290 290–300 |
0,38 0,52 0,62 0,76 |
0,392 0,519 0,64 0,764 |
0,37 0,50 0,59 0,72 |
0,011 0,015 0,015 0,020 |
0,37 0,5 0,59 0,72 |
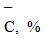
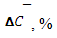
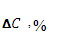
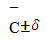
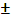 0,02
0,02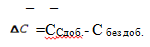
Величину 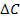 находят как корень квадратный из суммы квадратов стандартных отклонений значения ССдоб.- С без доб.
находят как корень квадратный из суммы квадратов стандартных отклонений значения ССдоб.- С без доб.
Таблица 2
Сопоставление результатов определения серы рентгеноспектральным и химическим методами
|
Пределы отбора фракций, 0С |
Найдено, % |
Пределы отбора фракций, 0С |
Найдено, % |
||
|
Рентгеноспектральным методом |
Химическим методом |
Рентгеноспектральным методом |
Химическим методом |
||
|
270–280 280–290 290–300 310–320 320–330 330–340 |
0,519 0,62 0,76 1,0 1,22 1,40 |
0,525 0,66 0,75 0,95 1,15 1,30 |
340–350 390–400 400–420 430–440 460–470 |
0,57 1,81 1,75 1,82 2,06 |
1,45 1,72 1,70 1,84 1,98 |
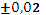
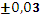

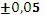
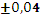
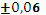
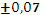
Для эксперимента отбирали 11 проб 10-градусных фракций зевардинской нефти. Из каждой пробы готовили два раствора, добавляя в каждый внутренний стандарт. Растворы заливали в две кюветы, испаряли растворитель и в каждом излучателе независимо дважды измеряли интенсивности аналитической и сравниваемой линий [7]. Такое планирование эксперимента позволило разложить суммарную погрешность методики на составляющие:
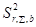 =
= 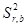 +
+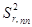 +
+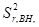
где 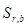 – относительное стандартное отклонение, определяемое нестабильностью аппаратуры в течение короткого промежутка времени и статистической природой распределения квантов во времени и пространстве;
– относительное стандартное отклонение, определяемое нестабильностью аппаратуры в течение короткого промежутка времени и статистической природой распределения квантов во времени и пространстве; 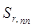 – величина, обусловленная неоднозначностью условий приготовления излучателей;
– величина, обусловленная неоднозначностью условий приготовления излучателей; 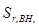 – величина обусловленная неоднозначностью введения внутреннего стандарта. При дисперсионном анализе погрешности [8] получили значения =0,7 % и =1,5 %. Погрешность приготовления излучателя незначима на фоне воспроизводимости измерения интенсивности SKα-линии. Суммарная погрешность методики
– величина обусловленная неоднозначностью введения внутреннего стандарта. При дисперсионном анализе погрешности [8] получили значения =0,7 % и =1,5 %. Погрешность приготовления излучателя незначима на фоне воспроизводимости измерения интенсивности SKα-линии. Суммарная погрешность методики 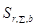 характеризуется относительным стандартным отклонением 1,64 %.
характеризуется относительным стандартным отклонением 1,64 %.
Заключение. Результаты этих исследований показывают, что основной вклад в погрешность на стадии подготовки пробы вносит неоднозначность введения внутреннего стандарта в пробу. Поэтому для повышения точности результатов РСФА целесообразно от каждой пробы брать по две навески и независимо вводить в них элемент сравнения, а затем из каждой навески готовить по одному излучателю. Если по каким-либо причинам использование двух излучателей затруднительно, целесообразно навески материала пробы после введения внутреннего стандарта слить вместе, тщательно смешать и из смеси приготовить один излучатель.
Правильность методики оценивали способом добавок (табл.1) и сопоставлением с данными химического анализа (табл. 2). Установили, что расхождение результатов сравниваемых методов определяется относительным стандартным отклонением 3,2 %. Учитывая, что воспроизводимость результатов химического анализа характеризуется относительным стандартным отклонением 3,6 %, с помощью F-критерия был сделан вывод об отсутствии значимых систематических погрешностей в результатах рентгеноспектрального анализа.
Предел обнаружения серы по разработанной методике, оценённый 30- критерию, составил 0,005 %.
За 8- часовой рабочий день разработанной методикой можно проанализировать 12–15 проб при двух параллельных взвешиваниях, включая препарирование излучателей, измерение интенсивностей и обработку результатов измерения.
Литература:
1. PrapuolenisA. A. Rapid determination of the iron content oils by X-ray fluorescence analysis//Radiochemical and Radio analyst Letters, 2007. V.№ 1. Р.11.
2. Lindeman L. Technical di analisitramitespectrometriadeiraggi X di fluorescenza// Riv. Combust. 1999. V.25 № 7–8. Р. 299.
3. Larson J. A., Short M. A., Bonfiglio S. An X- ray fluorescence technigue for the analysis of lead in gasoline // X- ray Spectrom. 1998. V. 3. № 3. Р. 125.
4. Marti Rosello J. Gonzales Escoda J., CarbeCarbe Pedro. Analysis de azufrepor fluorescence de rayos X // Oilgaz. 1996. V. 9. № 108. Р. 49.
5. Рентгенофлуоресцентный анализ. Применение в заводских лабораториях: Сб. науч. Тр./ Под ред. Эрхадра Х. М.: Металлургия, 1985. 107 с.
6. Смагунова А. Н., Базыкина Е. Н. Рентгенофлуоресцентный анализ растворов // Журн. аналит. Химии. 1985. Т. 40. № 5. С. 773.
7. Налимов В. В. Применение математической статистики при анализе вещества. М: Физматгиз, 1989. 463 с.
8. Климова В. А. Основныемикрометоды анализа органических соединений. М.: Химия, 1988. 104 с.
