





Синтезированы 5 разные производных азофенолов на основе реакции диазотирования и изучены химические свойства их галогенирования, алкилирования, а также реакции между изоцианатами.
В древние времена люди заботились о том, чтобы окрасить одежду и предметы домашнего обихода в красивые цвета. При религиозно-культовых обрядах, напротив, использовали отталкивающие расцветки. Во все времена окраска имела символическое значение, как это и сейчас выражается в цветах гербов и национальных флагов. 50-х годах 19 столетия органическая химия начало своё триумфальное шествие, одной из важнейших проблем, стоявших перед нею, являлось получение природных красителей синтетическим путем. [1]
Синтезированы 5 разных производных азофенолов на основе реакции диазотирования и изучены химические свойства их галогенирования, алкилирования, а также реакции между пропаргиловых эфиров и изоцианатов.
Из истории науки известно, что первые попытки связать химическое строение красителей и их светопрочность были сделаны, по-видимому, Гербардом. Он нашел атомы хлора, брома, сульфо- и карбоксильная группы замедляют выцветание; последняя особенно сильно. Имеет значение также и положение заместителя.
Наличие в молекуле красителя первичных аминогрупп обусловливает низкую светопрочность, а ацилирование (в особенности хлорированными красителями) повышает светопрочность [2].
В отличие от этого при галогенировании атака ароматического субстрата может осуществляться различными электрофилами. Свободные галогены CI2 и Br2, могут легко атаковать активированное ароматическое ядро. Для поляризации атакующей молекулы галогена необходимы катализаторы типа кислот Льюиса, такие как AICI3, с помощью которых в молекуле галогена появляется так называемый «электрофильный конец»: энергия, требующаяся для образования катиона CI+, очень велика [3]. Электронодонорные заместители в ароматическом кольце (-N=N-) ускоряют процесс и направляют галоген в орто- и пара-положение:
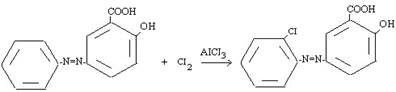
Азосоединении, вероятно, образует π-комплекс, например, с Br2, с которым затем взаимодействует кислота Льюиса. При бромировании в случае использования FeBr3 и других мало активных кислот Льюиса, образование катиона галогена идет в незначительной степени, а основной атакующей частицей является поляризованный комплекс, например:
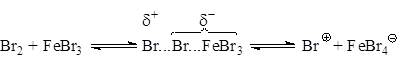
Катализатор, вероятно, поляризует связь Br-Br, способствуя образованию σ-связи между теперь уже электрофильным концом молекулы брома и атомом углерода кольца:
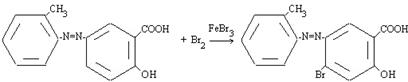
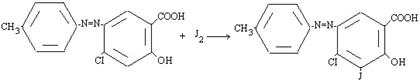
Изучены химические превращения производных азофенола: реакция йодирования протекает по механизму SE.
В гетеролитических реакциях замещения нуклеофильная частица часто предоставляет свою электронную пару для образования новой связи не сразу, а постадийно, то есть, реализуется механизм замещения с переносом электрона и образованием промежуточной анион-радикальных частиц.
Гомолитический путь замещения у насыщенного атома углерода осуществляется в основе по цепному радикальному механизму и характеризуется стадиями инициривания, роста и обрыва цепи [4]. К важнейшим реакциям этого типа относятся гомолитическое бромирование.
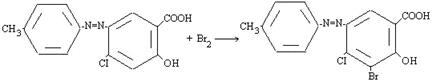
Нуклеофильные реакции замещения в зависимости от кинетических закономерностей разделяются на би- и мономолекулярные. Биомолекулярное замещение SN2 осуществляется через переходное состояние, в котором происходит одновременное образование новой и разрыв старой связи.
Нам известно, что изоцианаты являются высоко реакционноспособными соединениями. Они легко реагируют с соединениями, содержащими подвижные атомы водорода, например аминами и спиртами. Этот процесс широко используют в лакокрасочных составах для отверждения гидроксилсодержащих пленкообразователей. Взаимодействие изоцианатов с ОН-группами пленкообразователей протекает с достаточной скоростью уже при комнатной температуре. Этот процесс можно ускорить за счет введения катализаторов или повышения температуры. Скорость реакции зависит также от вида применяемого изоцианата [5].
Наука утверждает, что гидроксиазоарены являются ОН-кислотами. При ионизации их углубляется окраска, особенно в случаях о- и п- изомеров, где имеется прямое сопряжение с азогруппой [6]. Углубление окраски объясняется увеличением электронодонорных свойств заместителя при переходе от –ОН к –О-
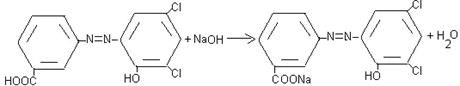
При действии разбавленной щелочейи на водные растворы красителя выделяются свободное основания, которое хорошо растворяется в некоторых растворителях (бензиловый спирт, феноксиэтанол) и используется в производстве чернильных паст.
А также изучены натриевая соль красителя при действии галогеналкана, синтезе ароматического сложного эфира. Реакция протекает по типу электрофильного замещения.
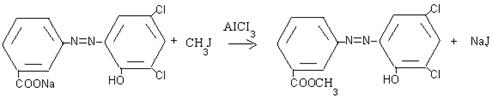
Обратимость реакции алкилирования по Фриделю-Крафтсу приводит к тому, что в системе одновременно идут все возможные реакции алкилирования и деалкилирования, причем затрагивается и мета-положение, так как алкильная- группа активирует все положения бензольного кольца, хотя и в разной степени.
Нами для получения соединения с биологически активными и красящими свойствами синтезированы пропаргиловые эфиры замещенных азо-гидроксибензолов. Эфиры получены взаимодействием замещенных азо-гидроксибензолов с хлористым или бромистым пропаргилом в присутствии поташа в среде ацетона при кипячении в течение 5–6 часов, по следующей схеме:
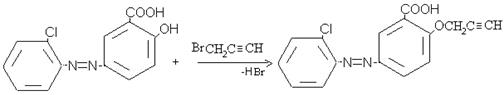
Полученные пропаргиловые эфиры представляет собой желтого цвета, со своеобразным специфическим запахом, хорошо растворимы в эфире, ацетоне, бензоле, этаноле, плохо растворимы в воде. Чистоту пропаргиловых эфиров контролировали ТСХ на незакрепленным слое AI2O3 IIстепени активности в системе бензол: бензол:этанол (10:1), бензол:гексан (10:1).
Полученные эфиры, благодаря наличию в их молекуле фрагментов азо-гидроксибензола и пропаргильной группы, проявляют антимикробные, противовоспалительные и красящие свойства.
Литература:
1. Бурятский Государственный Университет Курсовая работа по органической химии «Получение синтетических красителей реакцией азосочетания на примере синтеза 3-окси-4-карбоксиазобензола» стр. Улан-Уде, 2003г
2. Чекалин М. А., Пасет Б. В., Иоффе Б. А., Технология органических красителей и промежуточных продуктов, 2изд., Л., 1980.
3. Merkle K., Schiifer H., Pigment handbook, ed. By T. Patton, v. 3, 1973, p. 157–67.
4. Басодо Ф., Пирсон Р. Механизмы неорганических реакций, пер. с англ. М., 1971.,с. 28–32.
5. Б.Мюллер, У. Пот. Журнал «Лакокрасочные материалы и покрытия. Принципы составления рецептур». № 10, стр.20–25, 2006г
6. Нейланд. «Органическая химия» стр.434, Л., 1969г
